
MEN (Multipel Endokrin Neoplasi)
Resumé om MEN (Multipel Endokrin Neoplasi)
Hvad er MEN:
- Multipel Endokrin Neoplasi (MEN) er en gruppe sjældne, arvelige syndromer. De skyldes en medfødt genfejl, der markant øger risikoen for at udvikle tumorer (både godartede og ondartede) i flere af kroppens hormonproducerende (endokrine) kirtler.
MEN1 vs. MEN2:
- Man skelner primært mellem MEN1 (rammer oftest biskjoldbruskkirtler, bugspytkirtel og hypofyse) og MEN2 (rammer oftest skjoldbruskkirtel, binyrer og biskjoldbruskkirtler). MEN2 inddeles yderligere i MEN2A og MEN2B, som har forskellig grad af alvor og symptombillede.
Håndtering og kontrol:
- Selve genfejlen kan ikke helbredes, men tumorerne kan behandles. Håndteringen fokuserer på livslang, regelmæssig screening (blodprøver og scanninger) for at opdage og fjerne tumorer så tidligt som muligt, ofte før de giver symptomer.
Hvad er multipel endokrin neoplasi (MEN)
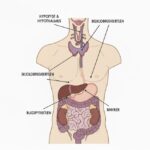
Multipel Endokrin Neoplasi, forkortet MEN, er ikke én specifik kræftsygdom. Det er en samlebetegnelse for arvelige syndromer, der skyldes en medfødt mutation (fejl) i et specifikt gen. Denne genfejl forstyrrer kroppens normale kontrolmekanismer og medfører en meget høj livstidsrisiko for at udvikle tumorer i flere forskellige endokrine kirtler.
De endokrine kirtler producerer hormoner, der regulerer kroppens funktioner. Når tumorer vokser i disse kirtler, kan de enten være godartede (benigne) eller ondartede (maligne/kræft). Uanset om de er ondartede eller ej, skaber de ofte problemer ved at overproducere hormoner, hvilket kan give en lang række symptomer.
At have et MEN-syndrom betyder, at man er i risiko for at udvikle tumorer flere steder og ofte i en yngre alder, end man ellers ville.
Årsager til MEN

MEN-syndromer er arvelige og skyldes en mutation i ét enkelt gen.
- MEN1 skyldes en mutation i MEN1-genet.
- MEN2 (både A og B) skyldes en mutation i RET-genet.
Syndromerne nedarves “autosomalt dominant”. Det betyder, at hvis én forælder har genfejlen, er der 50 % risiko for, at hvert barn arver den. Det betyder også, at syndromet rammer mænd og kvinder lige hyppigt. Tumorerne kan dog også opstå som følge af en ny (de novo) mutation, hvor man er den første i familien, der har den.
De forskellige typer af MEN

Det er afgørende at skelne mellem de forskellige typer MEN, da de involverer forskellige kirtler og har forskellig grad af risiko.
MEN type 1 (MEN1)
MEN1-syndromet kaldes også Wermers syndrom. Det er primært karakteriseret ved tumorer i tre kirtler (ofte kaldet “de 3 P’er”):
- Biskjoldbruskkirtlerne (Parathyroidea): Dette er den mest almindelige (ses hos over 90 %).
Tumorerne er næsten altid godartede (adenomer), men fører til overproduktion af parathyroideahormon (hyperparathyreoidisme). - Bugspytkirtlen (Pancreas): Her ses neuroendokrine tumorer (NETs), som kan være både godartede og ondartede.
De mest almindelige er gastrinomer (giver Zollinger-Ellisons syndrom med mavesår) og insulinomer (giver lavt blodsukker). - Hypofysen (Pituitary): Tumorer i hypofysen, oftets godartede.
Den hyppigste er prolaktinom (producerer for meget prolaktin).
Personer med MEN1 kan også udvikle tumorer i binyrerne (ofte godartede) og godartede tumorer i huden (lipomer).
MEN type 2 (MEN2)
MEN2 skyldes mutationer i RET-genet og opdeles i to hovedtyper:
MEN type 2A (MEN2A)
- MEN2A (Sipples syndrom) er den mest almindelige form af MEN2. Den er karakteriseret ved en kombination af:
- Medullær skjoldbruskkirtelkræft (MTC): Ses hos næsten alle (95-100 %). Dette er ofte den første tumor, der opdages.
- Fæokromocytom: En tumor i binyremarven, der producerer stresshormoner (adrenalin/noradrenalin). Ses hos ca. 50 % og kan forekomme i begge binyrer.
- Hyperparathyreoidisme: Tumorer i biskjoldbruskkirtlerne. Ses hos ca. 20-30 % (mindre hyppigt og ofte mildere end ved MEN1).
MEN type 2B (MEN2B)
- MEN2B er mere sjælden og generelt mere aggressiv end MEN2A. Den inkluderer:
- Medullær skjoldbruskkirtelkræft (MTC): Ses hos 100 % og opstår ofte meget tidligt i barndommen. Den er typisk mere aggressiv end ved MEN2A.
- Fæokromocytom: Ses hos ca. 50 %.
Det, der adskiller MEN2B, er, at man ikke udvikler sygdom i biskjoldbruskkirtlerne.
Til gengæld har man andre karakteristiske træk:
- Mukøse neuromer: Små, godartede knuder på tungen, læberne og i slimhinderne.
- Karakteristisk kropsbygning (marfanoid habitus) med lange, tynde lemmer.
- Forstyrrelser i mave-tarm-kanalen.
Symptomer på MEN
Symptomerne på MEN kommer ikke fra selve genfejlen, men fra de tumorer, der udvikler sig i de enkelte kirtler. Symptomerne afhænger derfor helt af, hvilken kirtel der er ramt, og hvilket hormon der overproduceres.
Ved hyperparathyreoidisme (MEN1 og MEN2A):
- Forhøjet kalk i blodet. Kan give træthed, knoglesmerter, forstoppelse, øget tørst og nyresten.
Ved fæokromocytom (MEN2A og MEN2B):
- Anfaldsvise symptomer pga. højt adrenalin. Hjertebanken, hovedpine, svedeture, angst og kraftigt forhøjet blodtryk.
Ved medullær skjoldbruskkirtelkræft (MEN2A og MEN2B):
- Giver ofte ingen symptomer tidligt. Senere kan mærkes en knude på halsen eller hævede lymfeknuder.
Ved NET i bugspytkirtlen (MEN1):
- Symptomer afhænger af hormonet. Gastrinomer giver svære mavesår. Insulinomer giver episoder med meget lavt blodsukker (svimmelhed, forvirring, svedeture).
Ved hypofysetumorer (MEN1):
- Prolaktinomer kan give nedsat sexlyst, infertilitet og mælkeproduktion (uanset køn). Andre tumorer kan give hovedpine eller synsforstyrrelser.
Diagnostik og udredning

Mistanken om MEN opstår typisk i tre situationer:
- En person diagnosticeres med en af de typiske tumorer (f.eks. MTC eller fæokromocytom).
- En person har tumorer i mere end én af de kirtler, der er associeret med MEN.
- Der er en kendt familiehistorie med MEN eller relaterede tumorer.
Den endelige diagnose stilles ved en gentest (en blodprøve), der analyserer MEN1-genet og RET-genet for mutationer.
Hvis en mutation bliver påvist, vil man straks igangsætte et screeningsprogram og tilbyde genetisk testning til relevante familiemedlemmer (forældre, søskende, børn).
Behandling og kontrol
Håndteringen af MEN er en livslang proces, der har to hovedformål:
- At behandle de tumorer, der allerede er opstået.
- At screene for og forebygge nye tumorer.
Behandling af tumorer
Behandlingen er typisk kirurgisk fjernelse af den eller de pågældende kirtler/tumorer.
- Hyperparathyreoidisme (MEN1/MEN2A): Operation, hvor man fjerner de forstørrede biskjoldbruskkirtler.
- Fæokromocytom (MEN2): Operation for at fjerne binyren. Dette kræver grundig medicinsk forberedelse for at blokere hormonernes effekt og undgå farligt højt blodtryk under operationen.
- NET i bugspytkirtlen (MEN1): Operation, hvis muligt. Medicin (f.eks. syrepumpehæmmere mod gastrinomer) bruges til at kontrollere symptomerne.
- Hypofysetumorer (MEN1): Behandles ofte først med medicin (f.eks. ved prolaktinomer), ellers operation.
Forebyggende operation (profylakse)
Ved MEN2 (især MEN2B) er risikoen for medullær skjoldbruskkirtelkræft 100 %, og den opstår ofte tidligt og aggressivt. Derfor anbefaler man stærkt en forebyggende (profylaktisk) fjernelse af hele skjoldbruskkirtlen – ofte allerede i det første leveår for børn med MEN2B-mutationen, og i den tidlige barndom for MEN2A.
Dette er en omfattende beslutning, men det gøres for at forhindre en kræftsygdom, der ellers er næsten uundgåelig.
Livslang screening
Den vigtigste del af at leve med MEN er det livslange screeningsprogram. Formålet er at opdage nye tumorer, før de bliver ondartede, eller mens de stadig er meget små og kan kureres.
Programmet skræddersys til den specifikke MEN-type og genmutation. Det består som standard af:
- Årlige blod- og urinprøver (måling af kalk, hormoner og tumormarkører som calcitonin).
- Regelmæssige billeddiagnostiske scanninger (MR, CT eller ultralyd) af de relevante organer (hals, brystkasse, mave, hjerne).
Det kan være en betydelig psykisk belastning at leve med en konstant overvågning og bevidstheden om den næste potentielle tumor.
Prognose og statistik

Prognosen for personer med Multipel Endokrin Neoplasi varierer dramatisk og afhænger specifikt af:
1) Hvilken type (MEN1, MEN2A eller MEN2B) man har
2) Om diagnosen stilles proaktivt gennem screening, eller først efter symptomer er opstået.
Læs mere…
MEN1
For MEN1 er den hyppigste tilstand (hyperparathyreoidisme) godartet og påvirker ikke i sig selv levetiden. Den afgørende prognostiske faktor er udviklingen af ondartede tumorer.
- Levetid: Flere studier indikerer en statistisk reduceret levetid sammenlignet med baggrundsbefolkningen.
- Et hollandsk studie har vist en median-overlevelse på 73 år, mod 80 år i kontrolgruppen.
- Årsager: Den primære årsag til nedsat levetid ved MEN1 er ondartede (maligne) neuroendokrine tumorer (NETs), særligt i bugspytkirtlen eller mave-tarm-kanalen, samt carcinoid-tumorer i brislen (thymus).
- Risiko: Risikoen for at udvikle en ondartet NET i bugspytkirtlen er betydelig over en livstid.
- Prognosen efter en sådan diagnose afhænger stærkt af tumorens stadium og aggressivitet ved opdagelsen.
MEN2A
Prognosen for MEN2A er generelt god, men er helt afhængig af håndteringen af medullær skjoldbruskkirtelkræft (MTC).
- MTC-overlevelse: Hvis MTC opdages og behandles tidligt, f.eks. ved forebyggende operation i barndommen, er overlevelsen næsten normal.
- 10-års overlevelsen for MTC, der er begrænset til skjoldbruskkirtlen, er over 90-95 %.
- Spredning: Hvis MTC derimod har spredt sig (metastaseret) til andre organer, falder 10-års overlevelsen til omkring 40-50 %.
- Fæokromocytomer: Disse tumorer i binyrerne er sjældent ondartede (under 5 %), men de er akut farlige, hvis de ikke opdages.
- Et udiagnosticeret fæokromocytom kan udløse en livstruende blodtrykskrise under f.eks. anden kirurgi.
MEN2B
Dette er historisk set den mest aggressive form af MEN, primært på grund af MTC, der opstår meget tidligt i livet og spreder sig hurtigt.
- Historisk prognose: Uden tidlig indgriben var median-levealderen for MEN2B kun omkring 21-25 år.
- Moderne prognose: Med aggressiv proaktiv behandling – det vil sige forebyggende fjernelse af skjoldbruskkirtlen inden for det første leveår – er prognosen forbedret dramatisk.
For alle MEN-typer gælder det, at en tæt og livslang opfølgning i screeningsprogrammet er den enkeltstående vigtigste faktor for at forebygge alvorlig sygdom og bevare en god prognose.
Arvelighed og genetisk vejledning

Da MEN er arveligt med 50 % risiko for videregivelse, er genetisk vejledning en central del af forløbet. Det giver mulighed for at drøfte konsekvenserne af at blive testet, herunder betydningen for familieplanlægning og forsikringsforhold.
At få konstateret MEN udløser ofte en kaskade af undersøgelser hos familiemedlemmer. Det kan være en svær proces at skulle informere slægtninge om, at de også kan være i risiko.
Komplementære og alternative tilgange

Mange søger at supplere den konventionelle behandling med livsstilsændringer. Det er vigtigt at fastslå, at ingen diæt, kosttilskud eller alternativ behandling kan ændre den underliggende genfejl eller fjerne risikoen for tumorer.
Nogle kan dog opleve, at en struktureret kost kan hjælpe med at håndtere symptomer fra hormonel ubalance (f.eks. ved insulinomer eller mavesår). Ligeledes kan afspændingsteknikker og mindfulness være værdifulde redskaber til at håndtere den stress og angst, der er forbundet med livslang screening og usikkerheden ved at leve med MEN. En supplerende tilgang bør altid drøftes med det behandlende team.
Konklusion

At modtage en diagnose med Multipel Endokrin Neoplasi (MEN) er en indgribende livsændring. Tilstanden adskiller sig fundamentalt fra sporadisk opstået kræft, da den defineres af en medfødt, genetisk disposition, der varer ved hele livet.
Den primære byrde ved MEN er ofte ikke kun de enkelte tumorer, men den psykologiske vægt af den konstante overvågning. Det er en proces præget af “vagtsom venten” og en tilbagevendende usikkerhed ved hver screening, hvilket kan være vanskeligt for omgivelserne at forstå.
Den viden, gentesten giver, er imidlertid også det stærkeste forsvar. Den tillader en proaktiv tilgang, hvor man kan være på forkant. For MEN-syndromerne er forskellen på en håndterbar tilstand og en livstruende sygdom næsten udelukkende et spørgsmål om timing.
Formålet med håndteringen er ikke at kurere den underliggende genfejl, men at forblive et skridt foran de tumorer, den kan forårsage. Gennem specialiseret screening og rettidig indgriben er målet at omdanne en genetisk risiko til en kronisk tilstand, der kan håndteres aktivt.
Links
[1] Multiple endokrine neoplasier (Patienthåndbogen på sundhed.dk)
- Indhold: En central dansk patientinformation, der definerer MEN og beskriver de forskellige typer (MEN1, MEN2A, MEN2B), de involverede kirtler, symptomer og behandlingsprincipper.
[2] Multipel Endokrin Neoplasi 1 (MEN1) (Rigshospitalet, ej dateret)
- Indhold: Specifik information fra Rigshospitalet om MEN1-syndromet, herunder de typiske tumorer i biskjoldbruskkirtler, bugspytkirtel og hypofyse samt det anbefalede kontrolprogram.
[3] Multipel Endokrin Neoplasi 2 (MEN2) (Rigshospitalet, ej dateret)
- Indhold: Specifik information fra Rigshospitalet om MEN2, der dækker tumorer i skjoldbruskkirtlen (MTC), binyremarven og biskjoldbruskkirtlerne samt det livslange kontrolforløb.
[4] Årsager til skjoldbruskkirtelkræft (Kræftens Bekæmpelse, 2025)
- Indhold: Beskriver den arvelige sammenhæng for medullær thyreoidea cancer (MTC), specifikt koblingen til MEN2-syndromet og mutationer i RET-genet.
[5] Multiple Endocrine Neoplasia (MEN) (Cleveland Clinic, 2022)
- Indhold: En omfattende international oversigt, der dækker diagnostiske kriterier, genetisk testning og symptomer for både MEN1 og MEN2.
[6] Multiple endocrine neoplasia (MedlinePlus Genetics, NCI, 2017)
- Indhold: En autoritativ amerikansk kilde, der grundigt gennemgår de genetiske årsager (MEN1- og RET-generne), arvelighed og de forskellige typer af MEN-syndromet.
❤
Hvad du læser på Jeg har Kræft er ikke en anbefaling. Søg kompetent vejledning.
Forfatterinfo og professionelt grundlag

Samtlige artikler på dette site er udarbejdet og valideret af undertegnede, Hanne Kjær Uhlig. Jeg er uddannet sygeplejerske (1975, med klinisk erfaring frem til 2013) og cand.arch. (1983, med speciale i industriel design), samt underviser i en årrække på DTU (Danmarks Tekniske Universitet).
Efter tabet af min mor til kræft i 2000 og min egen kræftdiagnose i 2024, stiftede jeg dette non-profit informationssite Jeg har Kræft.
Målet er at bruge min analytiske og akademiske systematik til at bringe overblik, sikkerhed og videnskabelig dokumentation ind i feltet for integrativ, komplementær og alternativ kræftbehandling. Samtidig benyttes min sundhedsfaglige erfaring til at gøre artikler patientnære – og vedkommende.
Artiklens kendetegn:
- Klinisk og personlig ballast: Skabt ud fra en kombination af årtiers erfaring som sygeplejerske og egne oplevelser som patient og pårørende.
- Videnskabelig systematik: Indholdet bygger på systematisk research af medicinske databaser samt kliniske forsøg. Artiklerne er konsekvent underbygget med kildehenvisninger under Links.
- Uafhængigt non-profit projekt: Driften sikres via frivillige bidrag og medlemskaber gennem Støtteforeningen Jeg har Kræft. Sitet er fuldstændig uafhængigt af kommercielle producentinteresser og arbejder udelukkende for at fremme kræftramtes livskvalitet.
- Støtteforeningens bestyrelse består af:
- Formand: Hanne Kjær Uhlig , sygeplejerske og cand.arch.
- Kasserer: Jens Ottosen-Støtt, jurist
- Medlem: Thomas Kjær Uhlig, speciallæge, øjensygdomme
- Medlem: Martin Kjær Uhlig, cand.merc.aud.
Fællesskab: Bliv medlem af Facebookgruppen: Jeg har Kræft – Hvad kan jeg gøre?
❤
Hvad du læser på Jeg har Kræft er ikke en anbefaling. Søg kompetent vejledning.